ADRENALS: DISORDERS OF THE ADRENALS
- See 2011 CUA Incidental Adrenal Mass Gudieline Notes
Disorders of Increased Adrenal Function
Hypercortisolism (Cushing Syndrome)
- Hypothalamus-Pituitary-Adrenal (HPA) Axis
- Hypothalamus
- Produces corticotropin-releasing hormone (CRH)
- Corticotropin-releasing hormone (CRH)
- Function:
- Acts on the corticotropic cells of the anterior pituitary to make ACTH
- Secretion
- Under tight control of the hypothalamic suprachiasmatic nucleus
- Follows circadian patterns.
- The highest level of cortisol in healthy subjects is detected in the mornings, and the nadir is observed at approximately 11 PM.
- Even small perturbations of this physiologic rhythm are considered pathologic
- The highest level of cortisol in healthy subjects is detected in the mornings, and the nadir is observed at approximately 11 PM.
- Function:
- ACTH
- Function:
- Stimulates production of glucocorticoids and androgens by the adrenal cortex
- Plays a critical role in maintaining adrenal cortical vitality.
- Without ACTH (e.g., when its secretion is suppressed by exogenous steroid intake), all but the mineralocorticoid(aldosterone)-producing cells of the adrenal cortex atrophy
- Secretion:
- Stimulated by:
- CRH (most important)
- Oxytocin
- Vasopressin
- Stimulated by:
- Function:
- Glucocorticoids act on to the hypothalamus and pituitary to inhibit production of CRH and ACTH (negative feedback)
- Patients with hypercortisolism may be at higher risk for post-adrenalectomy adrenal insufficiency than patients with non-cortisol secreting adrenal pathologies, because functionality of the contralateral gland may be suppressed
- Stress, whether physiologic or psychologic, appears to be the most important variable in modulating activity of the HPA axis
- Hypothalamus
- Hypercortisolism 2ry to excessive production of glucocorticoids by the adrenal cortex is defined as Cushing syndrome
- Pathophysiology
- Causes categorized into 3 main groups: exogenous vs. ACTH-dependent vs. ACTH-independent
- Exogenous
- Most common cause of hypercortisolism in patients of the Western world
- Can cause virilization, including hirsutism, but should not elevate ketosteroid levels
- ACTH-dependent (endogenous)
- 85% of cases of endogenous Cushing syndrome
- Results from an increased serum ACTH level
- Caused by pathology extrinsic to the adrenal gland:
- Primary pituitary pathology (also known as Cushing disease)
- Most common (80%) cause of ACTH-dependent hypercortisolism
- Ectopic ACTH production
- Nearly always malignant; the most common associated malignancies are bronchial carcinoid, small cell lung cancer, and less often pheochromocytoma
- Ectopic CRH syndrome
- Extremely uncommon; bronchial carcinoma is the most common cause
- Primary pituitary pathology (also known as Cushing disease)
- ACTH-independent (endogenous)
- 15% of cases of endogenous Cushing syndrome; relatively rare
- Cause by pathology intrinsic to the adrenal gland
- Results from unregulated overproduction of glucocorticoids by the adrenal(s), either unilateral neoplasm or rarely, bilateral disease
- Exogenous
- Causes categorized into 3 main groups: exogenous vs. ACTH-dependent vs. ACTH-independent
- Clinical characteristics
- See Table 65-2 for Primary Effects of Glucocorticoids
- Classic symptoms of hypercortisolism, such as central obesity, moon facies, buffalo hump, facial plethora, menstrual disturbances, hirsuitism, proximal muscle weakness, easy bruisability, and abdominal striae, are nonspecific.
- Cushing syndrome also results in systemic symptomatology, such as dyslipidemia, insulin resistance, and hypertension, similar to the highly-prevalent metabolic syndrome

Source: Wikipedia
- Urologogical complications of Cushing's syndrome
- Erectile dysfunction, decreased libido
- Hypogonadal hypogonadism
- Relatively common in men with Cushing syndrome
- Consider initiating a hypercortisolism workup in men with libido or erectile problems, low testosterone, and low gonadotropin levels
- Urolithiasis
- Up to 50% of patients with Cushing syndrome exhibit urolithiasis
- Stone formers with cushingoid features also should receive a hypercortisolemia evaluation
- Subclinical Cushing syndrome
- Hypercortisolemia in the absence of an overt cushingoid phenotype
- Surgical indications for subclinical Cushing syndrome are still a matter of debate.
- Some argue that adrenalectomy should be performed only in patients who are potentially symptomatic and exhibit clinical signs, such as hypertension, obesity, glucose intolerance, or osteopenia. Others argue that surgery must be offered to all patients to prevent the sequelae of hypercortisolism.
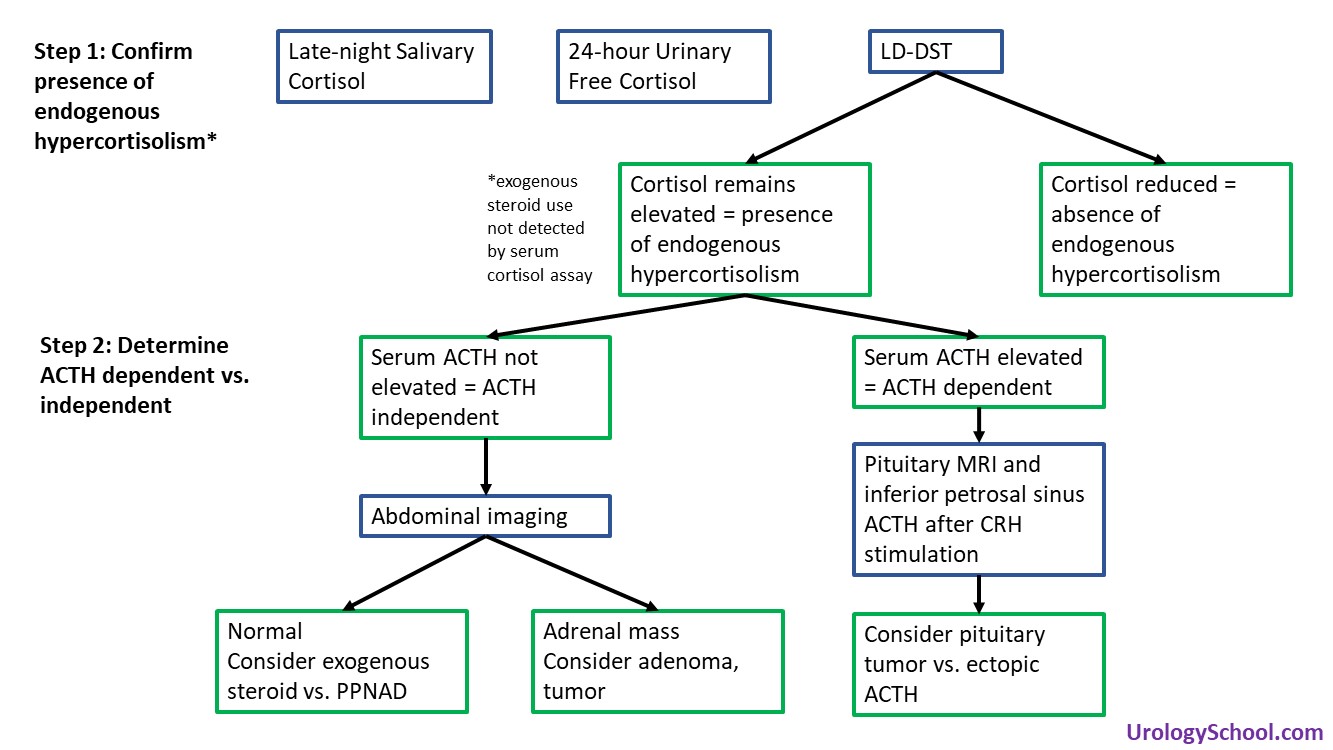
- Diagnosis and Evaluation
- The 3 tests performed most frequently are:
- Overnight low-dose dexamethasone suppression test (LD-DST) (sensitivity: 85-90, specificity: 95-99)
- Late-night salivary cortisol (sensitivity: 92-100, specificity: 93-100§)
- 24-hour urinary free cortisol (sensitivity: 80-98 specificity: 45-98)
- 2011 CUA Guidelines on Incidental Adrenal Mass recommend LD-DST
- Second-line tests:
- 2-day low-dose dexamethasone suppression test
- Midnight plasma cortisol testing
- Low-dose dexamethasone suppression test (LD-DST)
- Recommended in the evaluation of an incidental adrenal mass (2011 CUA Guidelines on Incidental Adrenal Mass)
- Determines the presence of endogenous hypercortisolism, not the cause
- To evaluate the patient’s glucocorticoid negative feedback system, a low-dose (1 mg) of dexamethasone is administered overnight followed by measurement of morning serum cortisol. The dose of dexamethasone administered is supraphysiologic and corresponds to 3-4x the level of physiologic glucocorticoids
- In patients without hypercortisolism, the dexamethasone acts on the corticotropic cells of the anterior pituitary, suppresses ACTH production, and thereby results in a reduction of serum cortisol levels.
- In patients with hypercortisolism due to endogenous causes, the dexamethasone fails to suppress cortisol production due to the relative insensitivity of pituitary adenomas to the inhibitory effects of glucocorticoid stimulation resulting in elevated serum cortisol levels despite dexamethasone.
- Exogenous steroid use cannot be ruled out with this test.
- Exogenous steroids, including that used for the test, are not detected by the serum cortisol assay.
- Pharmaceuticals That Affect Overnight Low-Dose Dexamethasone Suppression Testing for Cushing Syndrome
- Drugs that accelerate dexamethasone metabolism by induction of CYP3A4
- Phenobarbital
- Phenytoin
- Carbamazepine
- Primidone
- Rifampin
- Rifapentine
- Ethosuximide
- Pioglitazone
- Drugs that impair dexamethasone metabolism by inhibition of CYP3A4
- Aprepitant, fosaprepitant
- Itraconazole
- Ritonavir
- Fluoxetine
- Diltiazem
- Cimetidine
- Drugs that increase cortisol-binding globulin and may falsely elevate cortisol results
- Estrogens
- Mitotane
- LD-DST can yield as high as a 50% false-positive rate in women using oral contraceptives
- Contraceptives increase total (but not bioavailable) cortisol levels by raising the patient’s cortisol-binding globulin concentrations
- Late night salivary cortisol and midnight plasma cortisol demonstrate a perturbation, and in some cases complete disruption, of the diurnal variation of cortisol levels
- 24-hour urinary free cortisol
- May not be sensitive for subclinical Cushing syndrome, and the Endocrine Society recommends against it for metabolic evaluation of adrenal incidentalomas
- Drugs that increase urine free cortisol results
- Carbamazepine
- Fenofibrate (increase if measured by high-performance liquid chromatography)
- Some synthetic glucocorticoids (immunoassays)
- Drugs that inhibit 11β-hydroxysteroid dehydrogenase type 2 (licorice, carbenoxolone)

- Other conditions can stimulate the HPA axis and mimic Cushing’s syndrome
- Causes of hypercortisolism in the absence of Cushing’s syndrome:
- Some features of Cushing syndrome may be present:
- Morbid obesity
- Glucocorticoid resistance
- Poorly controlled diabetes mellitus
- Pregnancy
- Depression
- Alcohol dependence
- Unlikely to have any clinical features of Cushing syndrome
- Physical stress (hospitalization, surgery, pain)
- Malnutrition, anorexia nervosa
- Intense chronic exercise
- Hypothalamic amenorrhea
- Corticosteroid-binding globulin excess (increased serum but not in urine cortisol)
- After confirming hypercortisolism, serum ACTH is measured to distinguish ACTH-independent causes from ACTH-dependent causes
- Low serum ACTH
- Suggests ACTH-independent pathology
- Abdominal imaging is indicated to identify the adrenal source.
- If the adrenals are unremarkable on imaging, exogenous steroids as a cause of Cushing syndrome, or much less commonly, primary pigmented nodular adrenocortical disease (PPNAD) should be suspected.
- In PPNAD, the adrenal glands are normal in size and exhibit black or brown cortical nodules.
- If the adrenals are unremarkable on imaging, exogenous steroids as a cause of Cushing syndrome, or much less commonly, primary pigmented nodular adrenocortical disease (PPNAD) should be suspected.
- High serum ACTH
- Suggests pituitary source (Cushing disease) or ectopic ACTH syndrome
- Can be difficult to distinguish Cushing disease from ectopic ACTH syndrome because both pituitary and ACTH-producing tumours can be very difficult to localize with imaging.
- Direct measurements of ACTH in the inferior petrosal sinus, a downstream venous plexus that drains the pituitary, after CRH stimulation has become the gold standard approach for distinguishing ectopic ACTH production from Cushing disease.
- High-dose dexamethasone suppression testing was used in the past to differentiate pituitary and ectopic ACTH sources, but the value of the test is limited.
- The study is based on the principle that high enough doses of dexamethasone should suppress ACTH production by pituitary adenomas, whereas ectopic ACTH production continues despite the high-dose glucocorticoid administration
- Management
- ACTH-independent disease: ipsilateral adrenalectomy
- Medications that block enzymes of steroid synthesis (mitotane, metyrapone, aminoglutethimide trilostane, ketoconazole, etomidate) are used for bridging a hypercortisolism patient to surgery or when surgical intervention is not possible.
- Cushing disease (ACTH-secreting pituitary adenoma): trans-sphenoid surgical resection
- Bilateral adrenalectomy is most often recommended when at least one attempt to treat the primary tumor has failed. It is also necessary in rare instances when hypercortisolism is life-threatening and swift definitive treatment is mandatory. Lifelong mineralocorticoid and glucocorticoid replacement is required in all patients.
- Patients undergoing bilateral adrenalectomy are at risk (8-29%) for progressive growth of their pituitary adenoma, resulting in complications such as ocular chiasm compression, oculomotor deficiencies, and, rarely, a rise in intracranial pressure, resulting in the Nelson-Salassa syndrome (also known as Nelson syndrome), which is found in 8-29% of patients who have undergone bilateral adrenalectomy.
- When counseling patients regarding bilateral adrenalectomy for ACTH-dependent Cushing syndrome, the urologist must also warn of the rare possibility of residual, functioning adrenal tissue remaining after the procedure
- Ectopic ACTH production: resection of the ACTH-producing tumor.
- Primary tumor resection is possible in only 10% of patients.
- For patients with unresectable primary tumors or whose primary ACTH-producing tissue cannot be identified, bilateral adrenalectomy with lifelong replacement therapy is an excellent therapeutic option
Hyperaldosteronism
- Renin-Angiotensin-Aldosterone-System (RAAS)
- Under normal physiologic conditions, renin release is stimulated by (low-volume, low-salt state):
- Low renal perfusion pressure
- Increased renal sympathetic nervous activity
- Low sodium concentration sensed by the macula densa
- Renin then cleaves angiotensinogen to angiotensin I, which in turn is cleaved by angiotensin converting enzyme (ACE) to angiotensin II
- Angiotensin II
- Functions (2):
- Potent vasoconstrictor
- In kidneys, angiotensin II causes vasoconstriction of efferent and afferent arterioles, with stronger effect on efferent arteriole.
- Triggers the release of aldosterone
- Functions (2):
- Aldosterone
- Produced by:
- Zona glomerulosa of adrenal gland
- Production
- Stimulated by (3):
- Angiotensin II (most potent stimulator)
- Elevated serum potassium and decreased serum sodium
- ACTH (much less potent stimulator)
- The zona glomerulosa is the only region of the adrenal cortex that does not atrophy on pituitary failure
- Inhibited by:
- Atrial natriuretic peptide
- Stimulated by (3):
- Function:
- Increases sodium reabsorption and potassium secretion in the distal nephron.
- The increased sodium reabsorption increases total body volume.
- Increases sodium reabsorption and potassium secretion in the distal nephron.
- Produced by:

Source: Wikipedia
- Classification of hyperaldosteronism: primary vs. secondary
- Primary
- Aldosterone secretion is independent of the RAAS
- Renin levels are suppressed
- Can lead hypokalemia, hypomagnesemia, alkalosis, fluid depletion or retention, refractory hypertension, cardiac dysfunction and arrhythmias
- Hypernatremia does not occur because sodium reabsorption is accompanied by water uptake, thereby maintaining isotonicity.
- Most patients are normokalemic
- Although hypokalemia has been classically described as a common finding in primary aldosteronism, only 9-37% of newly diagnosed patients are hypokalemic
- Hypertension secondary to hyperaldosteronism is associated with an increased risk of end-organ damage compared with essential hypertension
- Causes (8):
- Bilateral hyperplasia (60%), also referred to as idiopathic hyperplasia;
- Clinically, patients with bilateral adrenal hyperplasia have less severe hypertension and are less likely to be hypokalemic compared with patients with aldosterone-producing adenomas
- Aldosterone-producing adrenal adenoma (35%)
- Unilateral adrenal hyperplasia (2%)
- Aldosterone-producing adrenal cortical carcinoma (<1%)
- Ectopic aldosterone-producing tumour (<1%)
- Familial hyperaldosteronism I (<1%)
- Aldosterone production is mediated by ACTH in familial hyperaldosteronism type I
- Familial hyperaldosteronism II (<1%)
- Familial hyperaldosteronism III (<1%)
- Secondary
- Elevated renin levels are the cause of elevations in aldosterone secretion
- Causes of elevated renin(4):
- Hypovolemia
- Juxtaglomerular cell tumour
- Renal artery stenosis
- Fibromuscular dysplasia
- Primary hyperaldosteronism
- Diagnosis and Evaluation
- Indications for primary hyperaldosteronism screening (9):
- Unexplained hypokalemia (spontaneous or diuretic induced)
- Hypertension with hypokalemia
- Adrenal incidentaloma with hypertension
- Resistant hypertension (3 or more oral agents with poor control)
- Early-onset hypertension (<20 years) or stroke (<50 years)
- Severe hypertension (≥160/≥110)
- Whenever considering secondary causes of hypertension (i.e., pheochromocytoma or renovascular disease)
- Evidence of target organ damage disproportionate to degree of hypertension
- Hypertension with family history of primary aldosteronism
- Primary hyperaldosteronism may be unmasked by diuretic-induced hypokalemia. The diagnosis is confirmed if 24-hour urinary aldosterone levels remain elevated after sodium loading (see below). After confirming diagnosis, a CT scan is done to localized the tumour.
- Labs
- Aldosterone-to-renin ratio (ARR)
- Used to screen for primary hyperaldosteronism.
- Involves measuring a morning (between 8-10 AM) plasma aldosterone concentration (PAC) and plasma renin activity (PRA).
- An ARR of > 20 (some suggest > 30) along with a concomitant aldosterone concentration > 15 ng/mL is indicative of hyperaldosteronism; standard thresholds have not been established due to laboratory variability.
- Before screening is initiated, hypokalemia should be corrected and all contraindicated medications discontinued.
- Although patients can continue the majority of anti-hypertensive agents during screening, potassium-sparing diuretics such as amiloride or triamterene, and especially mineralocorticoid receptor blockers such as spironolactone and eplerenone, alter the RAAS and will affect test results. These medications should be stopped approximately 6 weeks before testing.
- 50-70% of patients with a positive screening test will be diagnosed with primary aldosteronism following confirmatory testing. The majority of confirmatory tests evaluate the suppression of aldosterone after sodium loading.
- The underlying theory behind the sodium loading tests is that loading will decrease plasma renin and aldosterone production in patients without autonomous aldosterone secretion.
- The oral sodium loading test is conducted by administering a high-sodium diet for 3 days, followed by 24-hour urine measurements of aldosterone, sodium, and creatinine.
- The underlying theory behind the sodium loading tests is that loading will decrease plasma renin and aldosterone production in patients without autonomous aldosterone secretion.
- Imaging
- Cross-sectional abdominal imaging should be performed in all patients with primary aldosteronism who are potential surgical candidates.
- Radiographic characteristics of aldosterone-producing adenomas include the presence of a unilateral low-density non-enhancing lesion of < 10 Hounsfield units
- Lateralization of adrenal aldosterone-producing disease cannot be based on CT alone. Patients with confirmed primary aldosteronism should undergo adrenal vein sampling to establish lateralization when adrenalectomy is being considered. Exceptions include:
- Patients <40 years with a clear unilateral adrenal adenoma and normal contralateral adrenal gland on imaging
- Patients suspected of having an ACC
- Lateralization of adrenal aldosterone-producing disease cannot be based on CT alone. Patients with confirmed primary aldosteronism should undergo adrenal vein sampling to establish lateralization when adrenalectomy is being considered. Exceptions include:
- Genetic screening
- Given the rarity of familial primary hyperaldosteronism, genetic screening should not be performed in all patients. However, patients with a family history of primary aldosteronism, early age of onset (<20 years), or with a family history of cerebral vascular accidents at a young age should be considered for genetic testing
- Management
- Role of surgery depends on cause
- Surgically correctable causes of hyperaldosteronism (4)
- Aldosterone-producing adrenal adenoma
- Unilateral adrenal hyperplasia
- Ectopic aldosterone-secreting tumor
- Aldosterone-producing adrenal cortical carcinoma
- Non-correctable by surgery causes of hyperaldosteronism (4)
- Bilateral adrenal hyperplasia
- Familial hyperaldosteronism type I
- Familial hyperaldosteronism type II
- Familial hyperaldosteronism type III
- Surgically correctable causes
- In patients with confirmed lateralizing aldosterone secretion, adrenalectomy should be considered.
- Adrenalectomy Approach
- Given the small size of aldosterone-producing adenomas, the majority of patients are candidates for a laparoscopic adrenalectomy.
- In patients suspected of having hypersecretion of aldosterone associated with ACC, an open procedure may be recommended
- Non-correctable by surgery causes
- Medical treatment of primary aldosteronism in the form of mineralocorticoid receptor antagonists (spironolactone and eplerenone) is indicated in patients with non-surgically correctable subtypes and those who are not surgical candidates.
Pheochromocytoma
- Endocrine tumor that arises from the chromaffin cells of the adrenal gland
- ≈1/3 are familial cases
- 1-25% originate outside of the adrenal gland
- When it is found outside the adrenal gland, the tumor is called a paraganglioma.
- Extra-adrenal pheochromocytomas occur in:
- Organ of Zuckerkandl (located at bifurcation of aorta)
- Sympathetic chain
- Perivesical
- ≈5% of incidental adrenal masses will have a pheochromocytoma
- Currently, malignant pheochromocytoma can only be defined by the presence of clinical metastases.
- A number of pathologic criteria to differentiate benign from malignant disease have been proposed, but to date there is no criterion agreed on.
- Pathology
- Specific histopathological finding is zellballen, which are well defined nests of polygonal cells surrounded by fibrovascular stroma.
- Hereditary forms of pheochromocytoma
- In cases of VHL the risk of malignancy is low but pheochromocytoma is characterized as producing norepinephrine.
- Unlike pheochromocytomas in patients with VHL, MEN2 and NF1 predominantly produce epinephrine.
- Compared to sporadic cases, in familial syndromes, the pheochromocytomas are almost always bilateral and more frequently malignant. Clinical manifestations are similar.
Syndrome |
Clinical characteristics |
Risk of pheochromocytoma |
Risk of malignant disease |
Multiple endocrine neoplasia type 2A
|
|
50% |
3% |
Multiple endocrine neoplasia type 2B |
|
50% |
3% |
von Hippel-Lindau syndrome, type 2 |
HIPPPEEL
|
10-20% |
5% |
Neurofibromatosis type 1 |
|
1% |
11% |
Familial paraganglioma syndrome type 4 |
Carotid body tumors (chemodectomas) |
20% |
30-50% |
Familial paraganglioma syndrome type 1 |
Carotid body tumors (chemodectomas) |
20% |
<3% |

- Diagnosis and Evaluation
- History and Physical Exam
- History
- Clinical presentation
- See Table 65-7 for Clinical Manifestations of Pheochromocytoma
- > 20% of patients can be asymptomatic
- Classic triad:
- Headache
- Episodic sudden perspiration
- Tachycardia
- Can have heterogenous clinical behavior due to the variability in the amount and ratio of the different catecholamines (norepinephrine, epinephrine, dopamine) secreted:
- Norepinephrine-predominant tumours (e.g., patients with von Hippel- Lindau [VHL] syndrome) have hypertension and sweating because of norepinephrine's vasoconstricting action through the α adrenoreceptor
- Epinephrine-predominant tumours (rare, usually limited to adrenals or the organ of Zuckerkandl) have syncope or hypotensive episodes because of epinephrine’s vasodilatory action through the β2 receptor
- Agonist potency order:
- α1: epinephrine ≥ norepinephrine >> isoprenaline
- α2: epinephrine ≥ norepinephrine >> isoprenaline
- β1: isoprenaline > epinephrine = norepinephrine
- β2: isoprenaline > epinephrine >> norepinephrine
- β3: isoprenaline = norepinephrine > epinephrine
- Episodic hypertensive episodes may be triggered by events such as induction of anesthesia, labor and delivery, instrumentation and biopsy of the tumor, strenuous physical activity and consumption of tyramine rich foods, such as red wine, chocolate and cheeses.
- Another serious clinical presentation may result from catecholamine induced cardiomyopathy when patients present with congestive heart failure and cardiac arrhythmias.
- Clinical presentation
- History
- Labs
- Metanephrine Testing
- The enzyme phenylethanolamine-N-methyltransferase (PNMT), catalyzes the conversion of norepinephrine to epinephrine, is relatively unique to the adrenal medulla (the brain and organ of Zuckerkandl also express this enzyme).
- Localization of PNMT to the adrenal medulla explains why the gland is the primary source of systemic epinephrine, despite the presence of similar chromaffin cells elsewhere in the sympathetic nervous system
- Catecholamines (dopamine, norepinephrine, and epinephrine) are produced by pheochromocytomas in varying amounts; release of these compounds into the bloodstream is often paroxysmal
- Metanephrines are the methylated metabolites of catecholamines
- O-methylation of catecholamines is catalyzed by the COMT enzyme
- O-methylation of norepinephrine produces normetanephrine, whereas epinephrine’s methylation results in formation of metanephrine.
- Together, normetanephrine and metanephrine are known as metanephrines.
- O-methylation of catecholamines is catalyzed by the COMT enzyme
- The conversion of catecholamines to metanephrines within pheochromocytomas is an uninterepted process. As such, measurement of plasma concentration of metanephrines is much more sensitive for detecting pheochromocytomas than the measurement of rises in plasma catecholamines, which may be paroxysmal.
- In the past, measurement of both urinary and serum catecholamine levels was the mainstay for evaluation of pheochromocytoma. However, these tests had moderated sensitivity and specificity and have been largely replaced by measurements of levels of metanephrines.
- Measurement of urinary catecholamines, nevertheless, is still recommended in conjunction with urinary fractionated (see below) metanephrine testing
- Measurement of plasma fractionated metanephrines and 24-hour urinary fractionated metanephrines and catecholamines are the mainstay biochemical tests to diagnose pheochromocytoma. The former is used primary in those with high index of suspicion with the latter for those with low index.
- 2011 CUA Incidental Adrenal Mass guidelines recommend screening with a 24-hour urinary fractionated metanephrines and/or catecholamines.
- 2014 [most recent as of October 2019] Endocrine Society Pheochromocytoma Guidelines recommend plasma free metanephrines or 24-hour urinary fractionated metanephrines
- 2019 AUA Update on Pheochromocytoma recommended initial screening with plasma free metanephrines. Urinary fractionated metanephrines are an alternative, with slightly lower diagnosis sensitivity.
- The term fractionated is used when the laboratory report details not only the amount of each compound type (e.g., metanephrines), but also the relative concentrations of each compound (e.g., normetanephrine and metanephrine).
- The blood sample
should be drawn after placing an intravenous cannula, dimming
the room lights and having the patient lay supine for 30 minutes
after minimizing any pain or anxiety.
- Prior to the blood draw,
patients should be counseled on avoiding caffeine for a minimum
of 24 hours - Acetaminophen can produce a false-positive result owing to cross reactivity in the [serum?] assay and should be stopped for at least 5 days before testing.
- Tricyclic antidepressants and phenoxybenzamine should also be stopped, because these have been shown to be responsible for false-positive results.
- Usual antihypertensive therapy can be continued.
- Although β-blockade can potentially result in a false-positive test result, the current recommendation is to stop the medication only on repeat testing
- Prior to the blood draw,
patients should be counseled on avoiding caffeine for a minimum
- The enzyme phenylethanolamine-N-methyltransferase (PNMT), catalyzes the conversion of norepinephrine to epinephrine, is relatively unique to the adrenal medulla (the brain and organ of Zuckerkandl also express this enzyme).
- Vanillylmandelic Acid (VMA) Testing
- VMA is the primary end metabolite of catecholamines
- The sympathetic nervous system lacks the ability to produce epinephrine (lacks PNMT enzyme) and therefore contributes to the serum level of only normetanephrine (from norepinephrine) but not metanephrine (from epinephrine). Indeed, > 90% of metanephrine (an epinephrine metabolite) and some 20% or more of normetanephrine (a norepinephrine metabolite) in the bloodstream are derived from the adrenal medulla (PNMT is present in the brain and organ of Zuckerkandl). Therefore the relative rise of VMA levels, the combined total end metabolite of norepienphrine and epinephrine, in the presence of a pheochromocytoma is much less dramatic than the rise seen in the levels of metanephrines (from epinephrine), and therefore, the sensitivity of urine VMA levels is low. However, the specificity of VMA is high, especially in nonfamilial cases.
- Metanephrine Testing
- History and Physical Exam
-
Oral clonidine testing
- Used to distinguish suspected pheochromocytoma vs. essential hypertension in patients with minimally elevated plasma catecholamines.
- Patients with suspected pheochromocytoma usually present with elevated plasma catecholamines. However, they can rarely present with normal or mildly elevated plasma catecholamines.
- The oral clonidine test can help distinguish whether the signs and symptoms and minimally elevated plasma catecholamines are related to pheochromocytoma vs. essential hypertension, oral clonidine test.
- Patients with essential hypertension will experience a significant drop in norepinephrine due to suppression of production by the sympathetic nervous system, while those with pheochromocytoma will not.
- Used to distinguish suspected pheochromocytoma vs. essential hypertension in patients with minimally elevated plasma catecholamines.
- Imaging
- 18F-FDG PET (fluorine-18 fluorodeoxyglucose positron emission tomography)
- Gold standard imaging modality for definitive staging in patients with pheochromocytoma.
- Superior test characteristics to CT, MRI, and metaiodobenzylguanidine (MIBG) scintigraphy.
- Better accuracy than 123I-MIBG in nearly all patients, especially for identification of metastatic disease.
- MIBG
- Utilizes a small-molecule analog of norepinephrine
- High specificity but low sensitivity for diagnostic disease identification.
- Useful modality when a suspected pheochromocytoma
cannot be localized or when metastatic disease is
suspected.- In the most common and urologically most relevant clinical scenario, a solitary adrenal mass on cross sectional imaging in the setting of a biochemical evaluation indicative of pheochromocytoma, MIBG or 18F-FDG PET may be safely be omitted because these functional studies only serve to confirm what is already known and do not alter management. However, MIBG or 18F-FDG PET imaging for large (>5 cm) tumors is likely prudent to assess for metastatic disease before surgery and thereby counsel the patient appropriately
- MRI
- Distinct low signal intensity on T1-weighted imaging
- High signal intensity on T2-weighted imaging.
- Distinct low signal intensity on T1-weighted imaging
- CT
- On unenhanced CT, pheochromocytomas are typically > 10 HU (mean ≈35 HU) given their rich vascularity and low lipid content. This can help differentiate them from lipid-rich adenomas
- If the lesion is not an adenoma,
an adrenal mass protocol CT with IV contrast allows for evaluation of tumor washout.
- Benign adrenal lesions wash out >50% on delayed imaging while pheochromocytoma, adrenocortical carcinoma and metastatic tumors do not.
- Pheochromocytomas usually measure greater than 10 HU on unenhanced CT and >100 HU on contrast imaging, and are often well circumscribed in appearance with or without necrotic or cystic elements. Nevertheless, any evaluation of an adenoma should still include testing for plasma free metanephrines to rule out pheochromocytoma.
- If the lesion is not an adenoma,
an adrenal mass protocol CT with IV contrast allows for evaluation of tumor washout.
- On unenhanced CT, pheochromocytomas are typically > 10 HU (mean ≈35 HU) given their rich vascularity and low lipid content. This can help differentiate them from lipid-rich adenomas

Pheochromocytoma (dark circular shadow near body center) localized by MIBG scintigraphy.
Front and back views also show radioiodine collection in thyroid (neck) and bladder (pelvis).
See corresponding CT below.
Source: Wikipedia


CT scan with enhancement demonstrating suspicous left adrenal mass.
See corresponding MIBG scan above.
Source: Wikipedia
- Other
- Genetic counselling
- Investigation for familial
syndromes is warranted in a patient younger than 50 years
with a significant family history of an extraadrenal pheochromocytoma
(hereditary paraganglioma syndrome), or bilateral
or multifocal tumors.
- If a mutation is identified, screening should also be offered to asymptomatic at risk family members.
- Investigation for familial
syndromes is warranted in a patient younger than 50 years
with a significant family history of an extraadrenal pheochromocytoma
(hereditary paraganglioma syndrome), or bilateral
or multifocal tumors.
- Genetic counselling
- Managment
- Pheochromocytoma is a surgical disease. Complete resection of the tumor is advised whenever possible
- There is no level 1 evidence exists regarding optimal preoperative or perioperative management
- General principles (4):
- Pre-operative cardiology or anesthesia consultation because of risk for cardiomyopathy
- Preoperative cardiac workup, including electrocardiography and echocardiography, and assessment of hypertension-induced end-organ dysfunction are indicated.
- Pre-operative medications (α-Blockade followed by β-blockade)
- Restoration intravascular volume
- Monitored bed post-operatively
- Pre-operative medications
- All patients with pheochromocytoma and an abnormal metabolic evaluation undergo preoperative catecholamine blockade, including patients who do not exhibit evidence of blood pressure elevation and lack classic symptomatology.
- Catecholamine release during intraoperative tumor manipulation can result in hazardous blood pressure elevation and cardiac arrhythmias.
- Recent data suggest that preoperative α-blockade may not be necessary in normotensive asymptomatic patients
- α-Blockade
- Helps in both hemodynamic and glucose control
- Phenoxybenzamine
- Most common α- blocker used for preoperative catecholamine blockade of pheochromocytoma.
- MOA: irreversible, non-selective α receptor blocker
- Intraoperative catecholamine surges typically do not override its actions, because reversal of the blockade is possible only through synthesis of new receptor molecules.
- Non-selective nature may lead to tachycardia and β-adrenergic blockade may be necessary
- Prolonged hypotension in the immediate postoperative period and central nervous system effects such as somnolence may be expected
- Newer selective and competitive α1-adrenergic blockers such as doxazosin, prazosin, and terazosin obviate the drug-induced need for β-blockade.
- Started 7-14 days before surgery.
- Can be started at 10 mg twice daily with a
stepwise increase of 10 to 20 mg every 2 to 3 days until a final
dose of 1 mg/kg if tolerated.
- During this time blood pressure checks should be conducted at least 3 times a day.
- The last dose of phenoxybenzamine is usually given on the night before surgery, and the next morning’s dose is withheld to minimize potentially prolonged hypotension after tumor resection.
- Can be started at 10 mg twice daily with a
stepwise increase of 10 to 20 mg every 2 to 3 days until a final
dose of 1 mg/kg if tolerated.
- If phenoxybenzamine not effective for blockade, start metyrosine
- MOA of metyrosine: blocks the biosynthesis of catecholamines by inhibiting the conversion of tyrosine to L-dopa
- Generally added for extensive disease with large increases in catecholamines.
- Acute hypertensive
attacks can also be treated with a short-acting alpha blocker
such as phentolamine.
- β-blockade
- Must be given with caution in patients with myocardial depression
- Should never be started before appropriate α-blockade
- In the absence of α-blockade, β antagonists cause a potentiation of the action of epinephrine on the α1 receptors, resulting in hypertension, owing to blockade of the arteriolar dilation at the β2 receptor. For this reason, selective β1 adrenoreceptor blockers, such as atenolol and metoprolol, are usually preferred.
- May be added when (2)
- Systolic blood pressure is <100 mmHg
- Tachycardia or
reflex tachycardia develops.
- Calcium channel blockers
- Some studies report that sole use of calcium channel blockers is sufficient for safe pheochromocytomas resection. This approach avoids the reflex tachycardia and postoperative hypotension that are seen with use of phenoxybenzamine. This strategy be reserved for patients who are normotensive with paroxysmal hypertension and a normal baseline blood pressure.
- Usually, preoperative calcium channel blockade for 2 weeks is sufficient.
- Restoration intravascular volume
- Most important component of preoperative management
- Most centers admit patients the day before surgery and initiate aggressive IV fluid resuscitation
- Monitored bed post-operatively
- In the immediate postoperative period, consider overnight ICU admission for active monitoring
- If phenoxybenzamine was used for preoperative α-blockade, hypotension is common, given the lasting effects of the agent. Moreover, in a high catecholamine state, α2-adrenoreceptor stimulation inhibits insulin release. The withdrawal of this adrenergic stimulus after tumor resection may result in rebound hyperinsulinemia and subsequent hypoglycemia
- Post-operative follow-up
- Repeat metabolic testing should be performed ≈2 weeks after adrenalectomy to document normalization of catecholamine levels
- Postoperative cross-sectional imaging is reasonable to document tumor resection and appropriate healing of the resection bed.
- Subsequent imaging should be guided by results of biochemical testing
- Annual biochemical follow-up is mandatory for all patients with resected pheochromocytoma.
- Lifelong screening for recurrence is recommended
- 10-year recurrence rates are as high as 16%.
- No consensus on follow-up protocols exists.
- In a patient with pesistent hypertension 2-3 months after adrenalectomy, residual tumor somewhere else in the body must be considered. The patient should initially have plasma free metanephrine levels measured. If this is abnormal, MIBG scan may be helpful in identifying the location of this lesion.
- Treatment of Hereditary Pheochromocytoma
- Given that for patients with MEN-2 and VHL, the risk of malignancy is low whereas the risk of bilateral disease is significant, partial cortical-sparing adrenalectomy has been advocated. This strategy is used to avoid lifelong hormonal replacement, with its associated morbidity
- Treatment of Malignant Pheochromocytoma
- Currently therapy for metastatic pheochromocytoma is largely palliative.
- Surgical metastasectomy of resectable disease is the standard of care
- Little evidence exists to demonstrate that it prolongs patient survival or is more effective for symptomatic relief than medical treatment with α/β-blockade and α-methyl-p-tyrosine
- Chemotherapy is primarily used in patients in whom MIBG therapy has failed or in those whose tumors do not demonstrate MIBG uptake on initial MIBG imaging studies.
- Treatment of Pheochromocytoma in Pregnancy
- In a late term pregnancy, the patient should be treated with alpha-adrenergic blockade with phenoxybenzamine until the fetus has reached maturity to manage the hypertension. At this point, she should undergo caesarean section and tumor resection in one operation. The patient should not undergo the stress of vaginal delivery.
Disorders of decreased adrenal function
Adrenal insufficiency (Addison’s disease)
- Pathophysiology
- In the Western world, the most frequent cause of primary adrenal insufficiency is autoimmune adrenalitis
- Any patient who has undergone ipsilateral partial or radical nephrectomy and is undergoing contralateral renal or adrenal surgery is at risk for a postoperative adrenal crisis. Obtaining old operative or pathology reports and examining cross-sectional imaging for the presence or absence of adrenal tissue are essential in this setting
- In a patient with suspected post-operative adrenal crisis, consider 2mg dexamethasone or 100mg hydrocortisone
- Given that aldosterone secretion by the adrenals does not depend primarily on ACTH, the zona glomerulosa continues to function appropriately in patients with secondary adrenal insufficiency. Mineralocorticoid deficiency is therefore present only in patients with primary Addison disease
- Diagnosis and Evaluation
- See Campbell's Table for Clinical Manifestations of Adrenal Insufficiency
- Diagnosis of primary adrenal insufficiency is primarily made on clinical grounds, with a high index of suspicion given a patient’s history, examination findings, and laboratory evaluation.
- Diagnosis is confirmed by measurements of morning serum cortisol and ACTH. Patients with primary adrenal insufficiency also exhibit abnormal aldosterone and renin levels.
- Confirmatory testing involves assessing the adrenal response to ACTH stimulation in the form of the corticotropin test.
- Management
- Adrenal hormonal repletion
Congenital Adrenal Hyperplasia
- See Pediatrics Disorders of Sexual Differentiation Chapter Notes
- Autosomal recessive disorder
- Characterized by low cortisol production due a metabolic enzymatic abnormality in the cholesterol-steroid biosynthesis pathway; associated with a deficiency of 21-hydroxylase in >95% of cases
- In the absence of negative feedback, ACTH production by the pituitary is increased, resulting in hyperplasia of the adrenal cortex and overproduction of adrenal androgens
Adrenal cortical carcinoma (ACC)
- Pathogenesis
- The cause of sporadic ACC remains unknown
- Syndromes associated with ACC (6):
- Li-Fraumeni syndrome
- Beckwith-Wiedemann syndrome
- Lynch syndrome
- Carney complex
- MEN-1
- McCune-Albright syndrome
- Diagnosis and Evaluation
- History and Physical Exam
- History
- Symptoms can be secondary to local or systemic disease burden and/or hypersecretion of adrenal hormones.
- ACCs associated with hypersecretion of adrenal hormones are characterized as being functional.
- Most ACCs will be functional at the time of presentation and the most common hormone secreted is cortisol
- ACCs associated with hypersecretion of adrenal hormones are characterized as being functional.
- Symptoms can be secondary to local or systemic disease burden and/or hypersecretion of adrenal hormones.
- History
- History and Physical Exam
- Laboratory
- Evaluating the functional status of adrenal tumors suspicious for ACC is essential, not only for making the diagnosis of ACC but also for consideration of postoperative cortisol replacement and the potential use of tumor-secreted hormones as markers during postoperative surveillance.
- When considering the functional status of a tumor that raises suspicion for ACC, glucocorticoid, mineralocorticoid, catecholamine, sexual steroid, and steroid precursor excesses should be evaluated
- Imaging
- In incidentally detected adrenal tumors, size is a relative indicator of malignancy
- Risk of malignancy based on tumour size:
- < 4 cm: 5%
- > 4 cm: 10%
- > 6 cm: 25%
- Given the relationship between size and malignancy, it is currently recommended that adrenal tumors > 4-6 cm be surgically excised
- ACCs tend to be larger than benign adrenal tumors, with an average size of 10-12 cm on presentation
- Risk of malignancy based on tumour size:
- Radiographic characteristics of ACCs on CT imaging (5):
- Irregular borders
- Heterogenous enhancement
- Increased enhancement (mean 39 HU) compared to adenoma (8 HU)
- Calcifications
- Necrotic areas with cystic degeneration
- MRI§
- Normal adrenal gland
- TI: uniform intermediate signal intensity that is slightly less intense than that of the liver and renal cortical tissue
- T2: difficult to distinguish from retroperitoneal adipose tissue because of the presence of intracellular lipid with the gland
- Myelolipoma
- T1: bright
- T2: intermediate
- ACCs
- T1: isointense relative to the liver or spleen
- T2: intermediate to increased intensity
- Marked contrast uptake on gadolinium-enhanced images
- Pheochromocytoma
- Classically, bright signal intensity on T2-weighted imaging (best seen on fat suppression sequences)—termed the “light bulb” sign—was believed to be diagnostic. It is now clear that this imaging characteristic is neither specific nor sensitive enough to secure a diagnosis and must be interpreted with caution
- Other
- Percutaneous needle biopsy is usually not performed before surgical excision owing to a clinically unacceptable risk of needle-tract seeding.
- In cases of surgically resectable disease, the information obtained from biochemical and radiographic evaluation should be enough to justify extirpation.
- Percutaneous needle biopsy is usually not performed before surgical excision owing to a clinically unacceptable risk of needle-tract seeding.
- Pathology
- Modified Weiss criteria (5):§
Nectrotic
ACC
Metastasizes
- Necrosis
- Abnormal mitoses
- Cytoplasm (clear cells comprising ≤25% of the tumor)
- Capsular invasion
- Mitotic rate > 5 per 50 high power fields
- Calculate: 2x mitotic rate criterion + 2x clear cytoplasm criterion + abnormal mitoses + necrosis + capsular invasion (score of 3 or more suggests malignancy)
- Traditional Weiss criteria:
- Abnormal mitoses
- Mitotic rate >5/hpf
- Diffuse Architecture of tumor cells
- Clear cells ≤25%
- Capsular/sinusoidal/venous invasion
- Furman grade (3 to 4)
- Necrosis
- Management
- Multimodal treatment, surgical resection, radiation therapy, and systemic chemotherapy are often necessary
- Mitotane, an oral synthetic derivative of the insecticide dichlorodiphenyltrichloroethane (DDT), is the most commonly used chemotherapeutic agent in the treatment of ACC
- Mitotane inhibits several enzymes of the adrenal cortex including the cholesterol side-chain cleavage enzyme (P450scc, CYP11A1), 11β-hydroxylase (CYP11B1), 18-hydroxylase (aldosterone synthase, CYP11B2), and 3β-hydroxysteroid dehydrogenase (3β-HSD) to a lesser extent
- Prognosis
- Despite aggressive surgical resection, adrenal carcinoma is associated with a high rate (60-80%) of recurrent disease
- Factors associated with recurrence-free survival
- Tumor size
- Nodal status
- T stage
- Functional activity
- Capsular invasion
- Local and systemic adjuvant therapy is often administered despite any clear evidence demonstrating improved survival
- Factors associated with overall survival
- Margins status (most important)
- Tumor size
- Nodal status
Metastases
- Metastatic disease to the adrenals is common
- In patients with a history of a previous malignancy, >50% of newly discovered adrenal lesions are metastatic; nevertheless, metabolic workup in these patients is recommended.
- Bilateral and bulky disease (>4 cm) is necessary to produce biochemical evidence of adrenal insufficiency
- Current imaging modalities supplemented by adrenal biopsy, when necessary, can frequently differentiate metastases from a primary adrenal tumor.
Adrenal adenomas
- Benign
- Most common tumour arising from the adrenal gland
- Incidence increases with age
- Diagnosis and Evaluation
- Vast majority (93%) are metabolically silent
- The essential evaluation of the small adrenal mass requires differentiating the nonfunctional benign adenoma from functional or malignant lesions
- Imaging
- Vast majority (93%) are metabolically silent

CT scan demonstrating right adrenal adenoma
Source: Wikipedia
- Management
- Functional adenoma
- Should undergo resection in acceptable surgical candidate.
- Non-functional adenoma
- Size of lesion and its growth characteristics dictate management as described above
- Adenomas that are initially metabolically inert are unlikely (<2%) to gain function.
- Despite this low rate of “metabolic transformation,” the most recent consensus statement by a panel of NIH-convened experts suggests that annual metabolic hormonal screening for the first 3-4 years after diagnosis is prudent
Oncocytoma
- Extremely rare
- Despite predominantly benign lesions, a proportion of lesions can exhibit malignant potential
- On imaging, adrenal oncocytic lesions do not possess the central stellate scar often seen in renal oncocytomas
- Because the diagnosis of adrenal oncocytic tumor is nearly always made on surgical resection, the evaluation and treatment of oncocytic lesions follow the same strategy as that of other adrenal masses
Myelolipoma
- Rare, benign, metabolically silent lesions
- Possess tissue components identical to healthy bone marrow
- In the majority of cases, diagnosis of myelolipoma can be made accurately on cross-sectional imaging
- Usual enhancment on CT is between -30 and -140 Hounsfield Units
- The NIH consensus panel on adrenal incidentaloma concluded that myelolipoma can be regarded as an exception to the mandatory metabolic workup of a newly discovered adrenal mass
- Classically asymptomatic myelolipomas are treated conservatively; surgery is indicated only for extremely large or symptomatic lesions.
Genglioneuroma
- Extremely rare benign neuroectodermal neoplasms
- Tend to occur in the young and are composed of ganglion and Schwann cells
- Tumors can grow extremely large and have a propensity to encase vessels without impinging on the vessel lumen
Adrenal cysts
- 4 types of adrenal cysts have been described: pseudocysts, endothelial cysts, epithelial cysts, and parasitic cysts
- 7% of adrenal cysts are associated with malignancy, all of which were pseudocysts; because of the known chance of associated malignancy, observation of adrenal cysts must be done with caution
- Although the majority of adrenal cysts are benign and nonfunctional, routine endocrinologic evaluation should be performed to exclude active lesions
Evaluation of adrenal lesions in urologic practice
- See 2011 CUA Incidental Adrenal Mass Gudieline Notes
- Adrenal incidentalomas are unsuspected adrenal masses >1 cm identified on cross-sectional imaging performed for unrelated causes
- ≈20% of adrenal incidentalomas are found to be potential surgical lesions
- See Table 65-15 for Characteristics of Incidental Adrenal Masses as Described in a Systematic Review of Published Series of Adrenal Incidentalomas
- Imaging
- Ultrasound
- Suboptimal imaging modality for detecting and characterizing adrenal lesions
- CT
- An unenhanced CT scan is the first test
- Most easily interpreted test for intracellular lipid and therefore can diagnose an adrenal adenoma in more than 70% of cases
- Low attenuation (<10 HU) on unenhanced CT corresponds to high intracytoplasmic lipid content and is diagnostic for an adrenal adenoma
- This cutoff has ≈70% sensitivity and 98% specificity for the diagnosis of adrenal adenomas
- ≈30% of adrenal adenomas exhibit an attenuation >10 HU on unenhanced CT owing to their lower lipid content
- These “atypical adenomas” are indistinguishable from non-adenomas on non-contrast CT density measurements alone
- If an unenhanced CT is not indicative for an adrenal adenoma, a contrast enhanced CT with washout should be obtained
- A proper adrenal CT study includes (3):
- Non-contrast 5-mm images through the adrenal
- Enhanced (1-minute post-bolus imaging)
- 15-minute washout imaging
- The diagnostic information from a single-phase enhanced CT scan for adrenal lesions is quite limited, as there is considerable overlap in post-contrast attenuation of adenomas and non-adenomas
- Delayed (washout) imaging indicative of adrenal adenoma
- Absolute percent washout > 60% ([Enhanced − delayed]/[Enhanced − unenhanced] × 100%)
- Relative percent washout (RPW) > 40% ([Enhanced − delayed]/ [Enhanced] × 100%)
- Lipid-poor adenomas possess identical properties to lipid-rich adenomas regarding their rapid loss (washout) of enhancement after CT contrast
- RCC metastases and HCC mets may exhibit washout characteristics similar to those of lipid-poor adenomas
- MRI
- Relies on its ability to accurately quantify the lesion’s lipid content (similar to adrenal imaging by CT)
- CT washout studies are considered the gold standard and is better than chemical shift MRI for identifying adenomas
- Functional imaging
- The role of functional imaging for the diagnosis of pheochromocytoma is limited, given that most pheochromocytomas can be accurately diagnosed with cross-sectional imaging and metabolic evaluation for catecholamines and their metabolites.
- Size and growth
- The incidence of benign adrenal adenomas increases with age
- Adrenal lesions in younger patients, even those < 4 cm, must be managed with greater caution than similar lesions in an older patient.
- Lesions >4 cm in older patients with significant comorbidities may be better served with observation than resection
- Masses >6 cm should be considered malignant until proved otherwise.
- Although management of masses between 4-6 cm is controversial, in otherwise healthy individuals, masses >4 cm should be resected
- Growth kinetics should be followed. The current recommendation is to resect masses that grow >1 cm; however, incidence of malignancy among these patients is low
- Indications for adrenalectomy as per 2011 CUA Incidental Adrenal Masses Guidelines (4):
- Masses ≥4cm
- ≥0.5-1cm growth during follow-up
- Hyperfunctioning mass
- Imaging findings concerning for pheochromocyoma/malignancy
- Adrenal biopsy
- Limited role for the following reasons:
- Modern imaging in the context of clinical characteristics affords superb diagnostic capabilities
- Histologically, adenomas cannot be reliably differentiated from adrenal carcinomas
- Adrenal biopsy is not without risk
- Should be pursued only when limitations of imaging have been reached and when the physician and patient are certain that the result of biopsy will influence management.
- Perhaps the biggest role for adrenal mass biopsy is in patients with primary malignancies that have potentially recurred in the adrenal gland and whose management will be affected by the biopsy results.
- Always exclude possibility of pheochromocytoma before biopsy
- Assessment of function of adrenal masses
- 2011 CUA Incidental Adrenal Masses Guidelines recommend
- Low-dose dexamethasone suppression test or 24-hour urinary cortisol to rule out hypercortisolism
- 24-hour urinary metanephrines and/or catecholamines
- Aldosterone-renin ratio in patients with hypertension
- Testing for Adrenal Sex Steroid Hypersecretion
- Adrenal androgen secretion is under control of ACTH and, like cortisol, exhibits circadian patterns.
- Hypersecretion of adrenal sex steroids by adrenal masses, especially incidentalomas, is exceedingly rare
- Routine testing of incidentalomas for sex hormones is currently not recommended
Summary of surgical indications for adrenalectomy (first 4 consistent with CUA Incidental Adrenal Mass Guidelines)
- Size > 4 cm (with exception of myelolipoma)
- Size increases > 1 cm on follow-up imaging
- Adrenal hyperfunction
- Mass with imaging findings that are suggestive of malignancy (e.g., lipid poor, heterogeneous, irregular borders, infiltrates surrounding structures)
- Extremely large and/or symptomatic cyst or myelolipoma
- Isolated adrenal metastasis (multidisciplinary decision making required)
- During renal surgery for renal cell carcinoma if:
- Adrenal abnormal or not visualized because of large renal tumor size on imaging
- Vein thrombus to level of adrenal vein
- Failed neurosurgical treatment of Cushing disease, necessitating bilateral adrenalectomy
- Select patients with ectopic adrenocorticotropic hormone (ACTH) syndrome, requiring bilateral adrenalectomy
- ACTH-independent macronodular adrenal hyperplasia (AIMAH)
- Primary pigmented nodular adrenocortical disease (PPNAD)
Questions
- What is the role of ACTH? Where is ACTH secreted from? What stimulates release of ACTH?
- How are the causes of hypercortisolism/Cushing’s syndrome categorized? Which is the most common cause in the Western world? Which does Cushing’s disease fall under?
- What are clinical manifestations of hypercortisolism?
- What are non-adrenal urologic manifestations of hypercortisolism?
- What is subclinical Cushing’s syndrome?
- What non-radiographic tests can be used to detect Cushing’s syndrome?
- Which form of hypercortisolism cannot be evaluated with the low-dose dexamethasone suppression test?
- After confirming hypercortisolism, how can you distinguish ACTH-dependent from ACTH-independent causes?
- List causes of hypercortisolism other than Cushing’s syndrome.
- What is the most potent stimulator of aldosterone secretion? What are other stimulators of aldosterone secretion?
- What is the categorization of causes of hyperaldosteronism? What lab test can be used to differentiate them?
- List 8 causes of primary hyperaldosteronism
- List 9 indications for primary aldosteronism screening?
- Which medications should be held prior to testing for hyperaldosteronism?
- What are the surgically correctable subtypes of hyperaldosteronism? What are the non correctable by surgery subtypes of hyperaldosteronism?
- What laboratory test do the CUA guidelines recommend to rule out primary hyperaldosteronism?
- How is laterality of primary hyperaldosteronism established? When should this not be performed?
- What are medical treatments for the non correctable by surgery subtypes of hyperaldosteronism?
- Where can extra-adrenal pheochromocytomas originate from?
- List clinical manifestations of a pheochromocytoma
- What laboratory test do the CUA guidelines recommend to rule out primary pheochromocytoma?
- What is the gold standard imaging for pheochromocytoma?
- List the hereditary forms of pheochromocytoma
- Describe the key aspects of pre- and post-operative management of pheochromocytoma
- Which layer of the adrenal cortex continues to function in patients with secondary adrenal insufficiency?
- List clinical manifestations of adrenal insufficiency
- List syndromes associated with increased risk of adrenal cortical carcinoma
- What percentage of adrenal tumours >4cm are malignant? >6cm?
- List radiographic characteristics of ACC on CT imaging
- What is the most common hormone secreted by ACC?
- What are the absolute and relative percent washout on CT suggestive of adrenal adenoma?
- What is the initial imaging of choice for adrenal adenomas? What is the gold standard imaging for adrenal adenomas?
- List indications for an adrenalectomy.
Answers
- What is the role of ACTH? Where is ACTH secreted from? What stimulates release of ACTH?
- Stimulate production of glucocorticoids and sex hormones
- Anterior pituitary
- CRH from the hypothalamus
- How are the causes of hypercortisolism/Cushing’s syndrome categorized? Which is the most common cause in the Western world? Which does Cushing’s disease fall under?
- Exogenous vs. Endogenous. Endogenous classified as ACTH-dependant vs. ACTH-independent
- Exogenous most common cause in Western world
- Cushing’s disease is an ACTH-dependant cause
- What are clinical manifestations of hypercortisolism?
- Central obesity, moon facies, buffalo hump, facial plethora, erectile dysfunction, decreased libido, menstrual disturbances, hirsuitism, proximal muscle weakness, easy bruisability, and abdominal striae
- What are non-adrenal urologic manifestations of hypercortisolism?
- Hypogonadal hypogonadism (negative feedback from glucocorticoids on pituitary and hypothalamus) and urolithiasis
- What is subclinical Cushing’s syndrome?
- Hypercortisolemia without overt clinical manifestations
- What non-radiographic tests can be used to detect Cushing’s syndrome?
- Low-dose desamethasone suppression test
- Late night salivary cortisol
- 24 hour urinary cortisol
- Which form of hypercortisolism cannot be evaluated with the low-dose dexamethasone suppression test?
- After confirming hypercortisolism, how can you distinguish ACTH-dependent from ACTH-independent causes?
- Serum ACTH
- List causes of hypercortisolism other than Cushing’s syndrome.
- What is the most potent stimulator of aldosterone secretion? What are other stimulators of aldosterone secretion?
- Angiotensin II
- ACTH and elevated serum potassium
- What is the categorization of causes of hyperaldosteronism? What lab test can be used to differentiate them?
- Primary vs. secondary
- Plasma aldosterone-renin ratio
- List 8 causes of primary hyperaldosteronism
- Bilateral hyperplasia
- Aldosterone-producing adrenal adenoma
- Unilateral adrenal hyperplasia
- Aldosterone-producing ACC
- Ectopic aldosterone-producing tumour
- Familial hyperaldosteronism I
- Familial hyperaldosteronism II
- Familial hyperaldosteronism III
- List 9 indications for primary aldosteronism screening?
- Which medications should be held prior to testing for hyperaldosteronism?
- What are the surgically correctable subtypes of hyperaldosteronism? What are the non correctable by surgery subtypes of hyperaldosteronism?
- Surgically correctable: aldosterone-producing adrenal adenoma, unilateral adrenal hyperplasia, ectopic aldosterone-secreting tumor, aldosterone-producing adrenal cortical carcinoma
- Not correctable by surgery: bilateral adrenal hyperplasia, familial hyperaldosteronism type I, familial hyperaldosteronism type II, familial hyperaldosteronism type III
- What laboratory test do the CUA guidelines recommend to rule out primary hyperaldosteronism?
- How is laterality of primary hyperaldosteronism established? When should this not be performed?
- Adrenal vein sampling; lateralization cannot be established on imaging alone
- Patients <40 years with a clear unilateral adrenal adenoma and normal contralateral adrenal gland on imaging or patients suspected of having an ACC
- What are medical treatments for the non correctable by surgery subtypes of hyperaldosteronism?
- Mineralocorticoid receptor antagonists such as spironolactone and eplerenone
- Where can extra-adrenal pheochromocytomas originate from?
- List clinical manifestations of a pheochromocytoma
- What laboratory test do the CUA guidelines recommend to rule out primary pheochromocytoma?
- What is the gold standard imaging for pheochromocytoma?
- List the hereditary forms of pheochromocytoma
- Describe the key aspects of pre- and post-operative management of pheochromocytoma
- Alpha blockade x7-14 days prior to surgery +/- deferred start of beta-blocker if patient develops tachycardia
- Restoration intravascular volume, consider admitting the day before surgery
- Post-op ICU admission to monitor for hypotension, hyperinsulinemia and resulting hypoglycemia
- Which layer of the adrenal cortex continues to function in patients with secondary adrenal insufficiency?
- List clinical manifestations of adrenal insufficiency
- List syndromes associated with increased risk of adrenal cortical carcinoma
- What percentage of adrenal tumours >4cm are malignant? >6cm?
- List radiographic characteristics of ACC on CT imaging
- What is the most common hormone secreted by ACC?
- Cortisol
- What are the absolute and relative percent washout on CT suggestive of adrenal adenoma?
- What is the initial imaging of choice for adrenal adenomas? What is the gold standard imaging for adrenal adenomas?
- List indications for an adrenalectomy.
References
- Wein AJ, Kavoussi LR, Partin AW, Peters CA (eds): CAMPBELL-WALSH UROLOGY, ed 11. Philadelphia, Elsevier, 2015, chap 65
- Daneshvar M, Bratslavsky G. 2019 AUA Update on Pheochromocytoma.